Drug safety officer
Regulatory affairs director
Quality assurance manager
Medical director
Pharmacovigilance lead
Compliance officer

This process is used when the organization becomes aware of an adverse event associated with its product — through spontaneous reports, clinical trials, post-market surveillance, published literature, or partner notifications — and must assess the event and submit a report to regulatory authorities. It applies when the event may meet criteria for expedited reporting (such as serious and unexpected adverse reactions) or periodic aggregate reporting, and when the assessment requires medical judgment and regulatory expertise. It is common when safety scientists, medical officers, and regulatory teams must coordinate within strict regulatory deadlines. Ideal for pharmaceutical companies, biotech firms, medical device manufacturers, and marketing authorization holders in any jurisdiction.
The adverse event reporting process typically involves safety intake staff who log the initial report, drug safety officers or safety scientists who assess seriousness, causality, and expectedness, medical reviewers who provide clinical judgment on the event, regulatory affairs specialists who prepare and submit regulatory reports, and quality assurance who ensures process compliance.
On-time regulatory submissions by identifying reportable events early and tracking every case against its filing deadline. Accurate seriousness and causality assessment because medical review is built into the processing flow rather than performed as a downstream check. Complete regulatory submissions that include all required data elements, reducing health authority queries and follow-up burden. Consistent event processing across all reporting sources and jurisdictions, ensuring no reportable event is missed or under-classified. Audit-ready case records that demonstrate compliance with ICH, FDA, EMA, and other applicable regulatory frameworks.
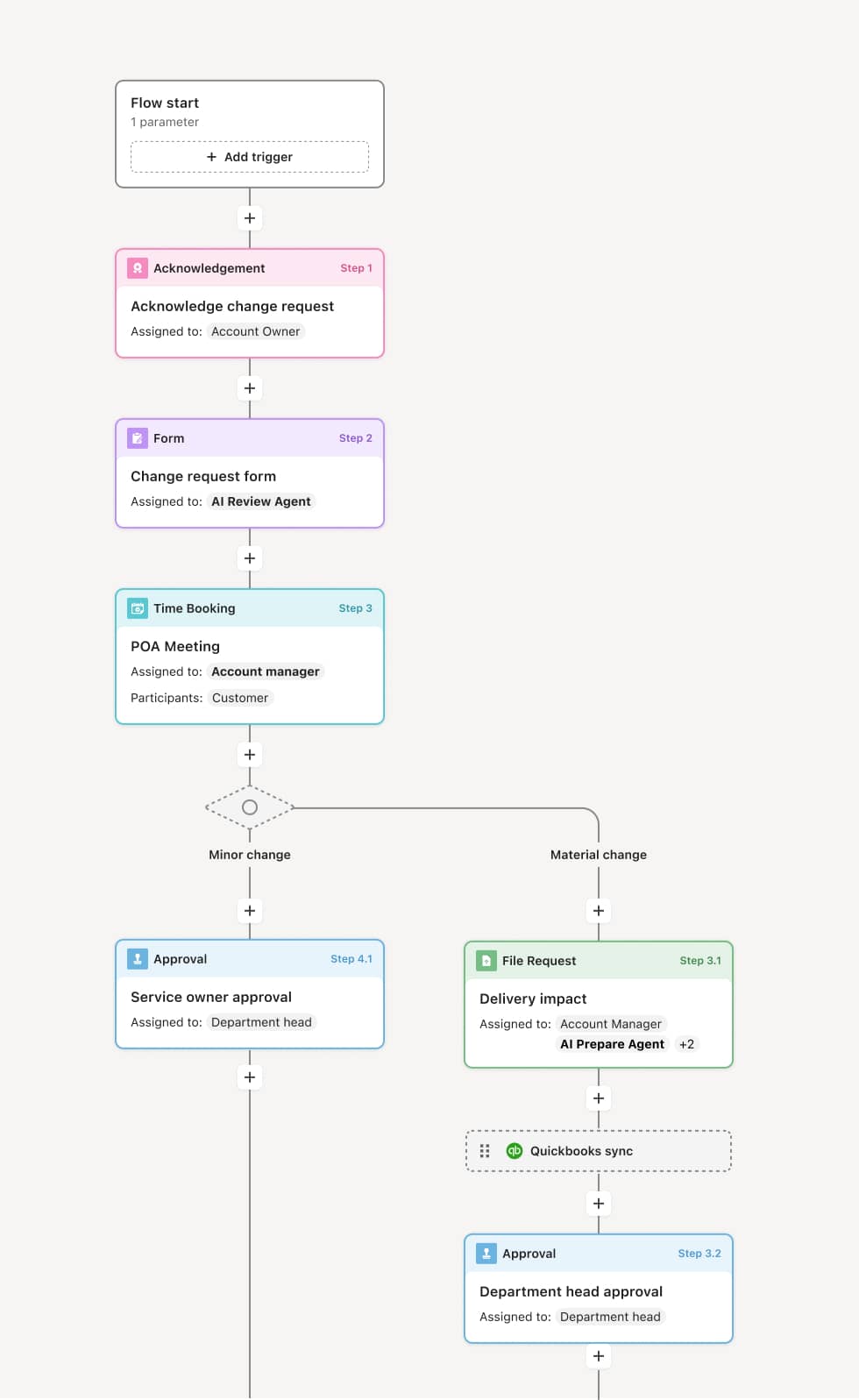
Your version of this process may vary based on roles, systems, data, and approval paths. Moxo’s flow builder can be configured with AI agents, conditional branching, dynamic data references, and sophisticated logic to match how your organization runs this workflow. The steps below illustrate one example.
Event identification and initial logging
The process begins when the organization becomes aware of an adverse event from any source. The event is logged with available details including the reporter, patient, product, event description, and outcome. An AI Agent can assist by extracting structured data elements from unstructured reports, identifying the product involved, and confirming that minimum case validity criteria are met.
Seriousness and expectedness assessment
A drug safety officer evaluates the event against regulatory criteria for seriousness — including death, hospitalization, disability, and other medically important conditions. The event is compared against the product’s reference safety information to determine expectedness. Serious and unexpected events are flagged for expedited 15-day reporting.
Medical and causality review
A medical reviewer assesses the clinical details of the event, evaluates the plausibility of a causal relationship with the product, and documents the clinical narrative. If additional information is needed from the reporter, a follow-up request is generated. An AI Agent may compare the event against the product’s known safety profile and similar historical cases to support the causality assessment.
Regulatory report preparation
Regulatory affairs prepares the adverse event report in the required format — typically an ICSR in E2B format for health authority submission. The report includes all required data fields, the medical narrative, causality assessment, and any supporting documentation.
Quality review and approval
The completed report undergoes quality review to verify data accuracy, completeness, and consistency with the source documentation. The report is approved for submission by the designated responsible person.
Submission and confirmation
The report is submitted to the applicable regulatory authorities through electronic gateways or designated channels within the required timeline. Submission confirmation is captured and the case enters the follow-up lifecycle for any additional information received.
This process commonly relies on inputs such as the adverse event report, patient medical history, product information, concomitant medications, and the reporter’s clinical observations. It may be triggered by a spontaneous report, clinical trial safety data, literature review, regulatory authority notification, or partner safety data exchange. Connected systems often include safety databases like Argus, ArisGlobal, or Veeva Vault Safety, E2B regulatory submission gateways, and clinical trial management systems.
Key decision points include whether the event meets minimum case validity criteria for processing, the seriousness classification that determines the reporting timeline, whether the event is expected or unexpected based on the product’s reference safety information, and which regulatory authorities require notification and within what timeframe.
Adverse events not recognized as reportable by frontline staff who receive initial reports, delaying case creation. Seriousness underassessed at initial triage, causing expedited cases to be processed on standard timelines. Medical review delayed due to competing priorities, compressing the window for quality review and regulatory submission. Multi-jurisdiction reporting requirements missed when the case is submitted to some but not all applicable authorities. Follow-up information not integrated into the original case in time for submission, requiring amendments and additional regulatory correspondence.
Orchestrates adverse event reporting from event identification through regulatory submission across safety, medical, regulatory, and quality teams in a single coordinated flow.
Routes cases based on seriousness and expectedness so expedited events receive immediate processing while standard cases follow periodic timelines.
AI Agents extract case data from incoming reports and flag missing elements, seriousness indicators, and unexpected events at the point of intake.
Tracks regulatory submission deadlines for every applicable jurisdiction, ensuring no filing window is missed.
Connects to safety databases and regulatory gateways like Argus, ArisGlobal, and Veeva Vault Safety so case processing flows directly into submission-ready formats.
Preserves the complete reporting record including source reports, assessments, submissions, and follow-up communications for audit, inspection, and signal detection.