IRB coordinator
Principal investigator
Research compliance officer
Institutional review board chair
Clinical research manager
Regulatory affairs specialist

This process is used when an active research study approaches its IRB-determined continuing review date and must be reassessed before the current approval period expires. It applies when the principal investigator must report on study progress, adverse events, protocol deviations, enrollment status, and any changes to the risk-benefit profile. It is common when IRB coordinators, investigators, and board members must coordinate within regulatory deadlines to prevent lapses in approval. Ideal for academic medical centers, contract research organizations, pharmaceutical companies, and any institution conducting human subjects research under IRB oversight.
The continuing review process typically involves principal investigators who submit progress reports and updated study documentation, IRB coordinators who track review schedules and manage submissions, IRB board members who evaluate the study’s ongoing risk-benefit profile and compliance, and research compliance officers who verify regulatory alignment and institutional policy adherence.
No lapses in IRB approval by tracking continuing review deadlines and ensuring submissions are completed before the approval period expires. Current risk-benefit assessment so the board evaluates the study’s ongoing justification based on the latest enrollment, adverse event, and protocol deviation data. Documented regulatory compliance with complete records of each continuing review cycle for accreditation and audit purposes. Reduced investigator burden by providing structured submission templates and clear requirements so continuing review materials are prepared efficiently. Proactive identification of study concerns because periodic review surfaces emerging safety signals, recruitment challenges, or protocol drift before they escalate.
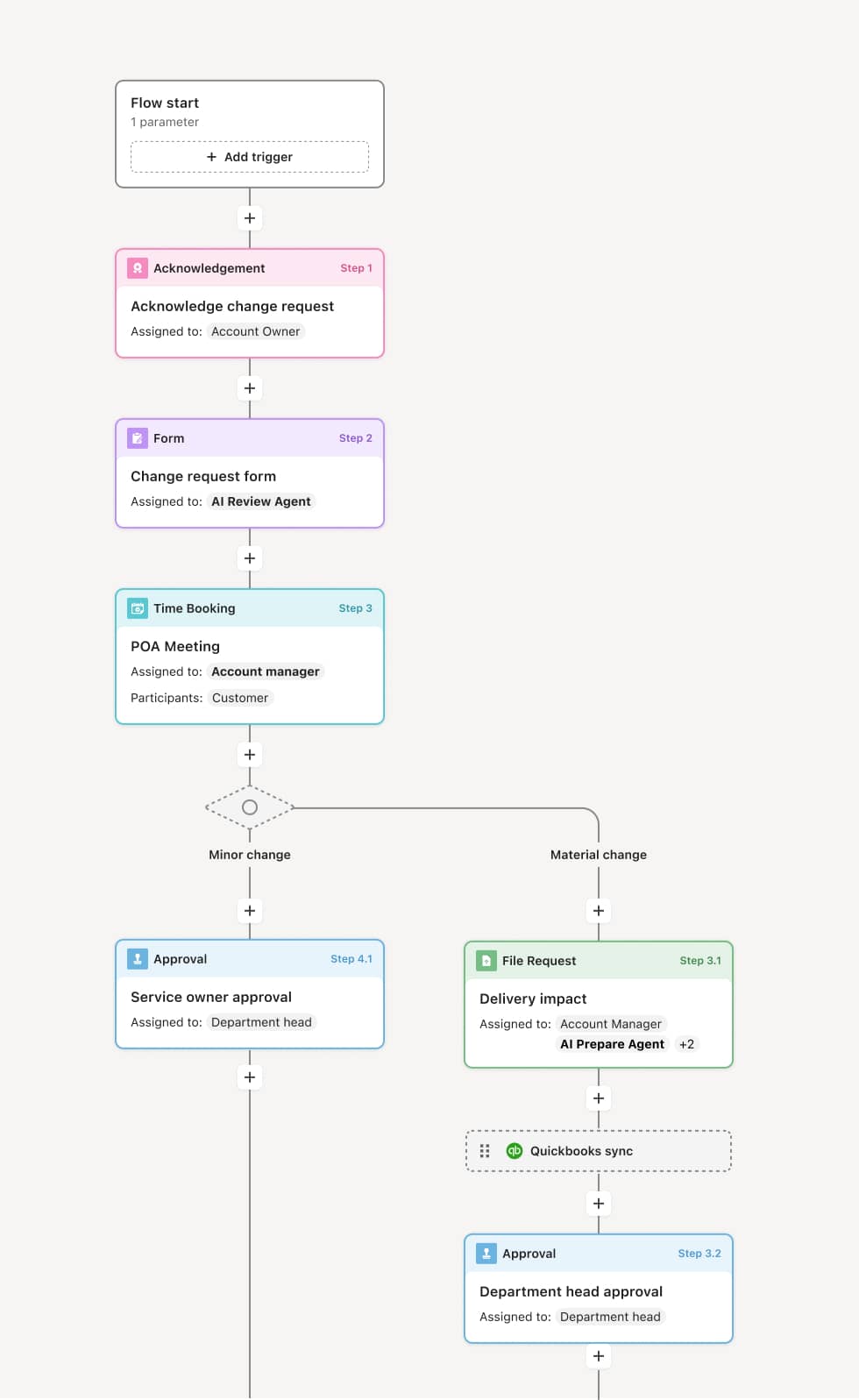
Your version of this process may vary based on roles, systems, data, and approval paths. Moxo’s flow builder can be configured with AI agents, conditional branching, dynamic data references, and sophisticated logic to match how your organization runs this workflow. The steps below illustrate one example.
Continuing review notification
The process begins when the IRB coordinator triggers the continuing review cycle, typically 30 to 60 days before the current approval period expires. The principal investigator is notified of the upcoming deadline and provided with the continuing review submission requirements. An AI Agent can assist by pre-populating the submission with known study data such as enrollment numbers, protocol version, and prior review outcomes.
Investigator submission
The principal investigator completes the continuing review report, including study progress, current enrollment, adverse events and safety reports, protocol deviations, changes to the risk-benefit profile, and any proposed modifications. Supporting documents such as updated consent forms, data safety monitoring reports, and publications are attached.
Administrative review
The IRB coordinator reviews the submission for completeness and accuracy, confirming that all required elements are present and that the study information is current. If the submission is incomplete, it is returned to the investigator with specific feedback. An AI Agent may validate the submission against the required checklist for the study’s risk level.
Board review and assessment
The submission is routed to the IRB board or designated reviewer based on the study’s risk classification. The reviewer assesses whether the study continues to meet ethical and regulatory standards, whether the risk-benefit ratio remains favorable, and whether participant protections are adequate. For full board studies, the review proceeds to a convened meeting.
Board decision
The board issues a decision — approval for a new review period, approval with modifications required, suspension, or termination. If modifications are required, the investigator must address them and resubmit before the new approval period is finalized.
Approval issuance and record preservation
Upon approval, a new approval letter is issued with the updated expiration date. The continuing review record — including the investigator’s report, board deliberation, decision, and any required modifications — is preserved for regulatory compliance and accreditation.
This process commonly relies on inputs such as the continuing review report, enrollment data, adverse event summaries, protocol deviation logs, data safety monitoring reports, and updated consent documents. It may be triggered by an IRB scheduling calendar, an automated deadline reminder, or a system alert from an electronic IRB platform. Connected systems often include electronic IRB platforms like iRIS or IRBManager, clinical trial management systems (CTMS), and institutional compliance databases.
Key decision points include whether the continuing review submission is complete and accurate, whether the study’s risk-benefit profile remains favorable based on current data, whether adverse events or protocol deviations require changes to the study or consent process, and whether the board approves continued research, requires modifications, or recommends suspension or termination.
Late continuing review submissions that risk a lapse in IRB approval and require suspension of research activities. Incomplete progress reports that delay board review because required safety data, enrollment numbers, or deviation logs are missing. Disconnected adverse event data when safety information is tracked in separate systems and not consolidated for the continuing review. Board scheduling conflicts that delay review beyond the approval expiration date. Investigator uncertainty about which modifications require board review versus administrative handling.
Orchestrates the full continuing review cycle from deadline notification through board decision across investigators, coordinators, and board members in a single coordinated flow.
Tracks continuing review deadlines proactively so submissions are triggered well before approval expiration, preventing lapses.
AI Agents pre-populate continuing review submissions with enrollment data, adverse event summaries, and prior review outcomes to reduce investigator preparation time.
Routes submissions based on study risk level so expedited reviews proceed quickly while full board studies follow appropriate deliberation timelines.
Connects to electronic IRB and CTMS platforms like iRIS, IRBManager, and clinical trial management systems so study data flows into the review process.
Preserves the complete continuing review record including investigator reports, board decisions, modifications, and approval letters for regulatory compliance and accreditation audits.