Quality assurance manager
Regulatory affairs specialist
Complaints coordinator
Product engineering lead
Post-market surveillance director
VP of quality

This process is used when the organization receives a complaint about a medical device from any source — healthcare providers, patients, distributors, or internal quality monitoring. It applies when the complaint involves a potential product defect, performance issue, adverse event, or injury associated with the device, and must be investigated and documented in compliance with FDA 21 CFR 820, ISO 13485, and Medical Device Reporting (MDR) requirements. It is common when customer-facing teams, quality, engineering, regulatory affairs, and senior management must coordinate to assess risk, investigate root cause, and determine regulatory reporting obligations. Ideal for medical device manufacturers, contract manufacturers, distributors, and any organization subject to medical device quality system regulations.
The complaint handling process typically involves customer-facing or post-market surveillance teams who receive and log the complaint, complaints coordinators who triage and assess initial risk, quality engineers who investigate root cause and assess product impact, regulatory affairs specialists who determine reporting obligations and prepare MDR submissions, and senior quality leadership who approve corrective actions and regulatory submissions.
Regulatory compliance by ensuring every complaint is investigated and documented in accordance with FDA and ISO requirements within mandated timeframes. Timely MDR submissions because reportable events are identified early in the investigation and regulatory affairs is engaged before filing deadlines. Thorough root cause investigation that connects complaint data to product design, manufacturing, and field performance to prevent recurrence. Reduced patient safety risk through prompt investigation and corrective action on complaints involving potential harm. Complete complaint history that supports post-market surveillance, trend analysis, and regulatory inspection readiness.
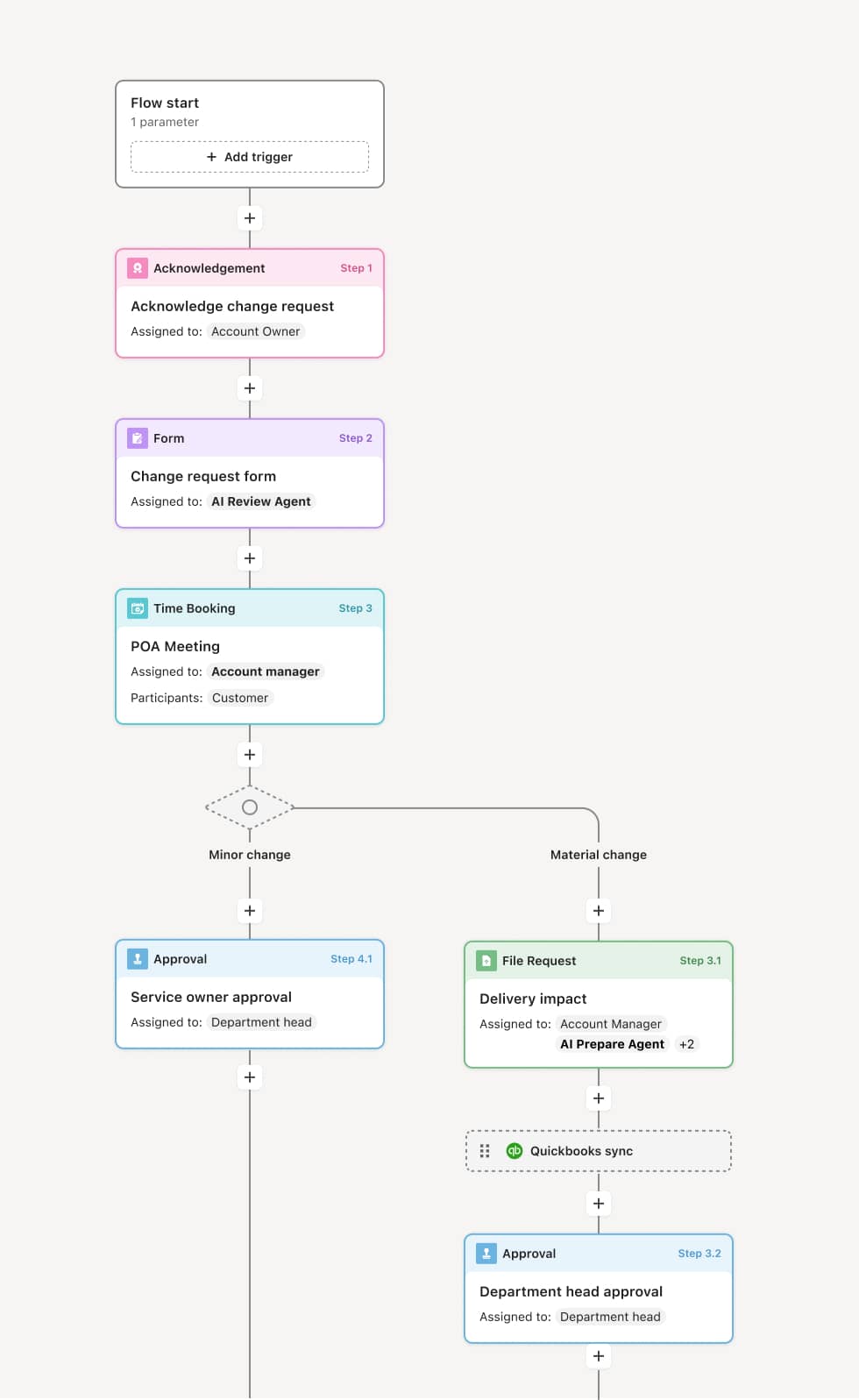
Your version of this process may vary based on roles, systems, data, and approval paths. Moxo’s flow builder can be configured with AI agents, conditional branching, dynamic data references, and sophisticated logic to match how your organization runs this workflow. The steps below illustrate one example.
Complaint intake and logging
The process begins when a complaint is received from any channel — customer call, field service report, distributor notification, or internal quality observation. The complaint is logged with the device identification, lot or serial number, description of the issue, complainant contact information, and any reported patient impact. An AI Agent can assist by parsing the complaint narrative to extract key data elements and flag language indicating a potential adverse event or reportable condition.
Initial risk assessment and triage
The complaints coordinator performs an initial risk assessment to classify the complaint by severity, patient impact, and potential regulatory reporting obligation. Complaints involving death, serious injury, or malfunction that could cause harm are flagged for expedited investigation and regulatory review. Lower-risk complaints follow the standard investigation timeline.
Investigation and root cause analysis
The quality engineering team investigates the complaint to determine root cause. Investigation activities may include review of manufacturing records, inspection of returned product, analysis of design specifications, review of similar complaints, and testing. An AI Agent may surface related complaints, CAPA records, and prior investigation findings to provide context for the current investigation. If the returned device is available, it is examined and documented.
Regulatory reporting determination
Regulatory affairs evaluates the investigation findings against MDR reporting criteria. If the event meets the threshold for a reportable event — such as a death, serious injury, or malfunction likely to cause harm — the MDR report is prepared and submitted to the FDA within the required timeframe. If the event does not meet reporting criteria, the determination and rationale are documented.
Corrective and preventive action assessment
Based on the investigation, the quality team determines whether corrective or preventive action (CAPA) is required. If a systemic issue is identified, a CAPA is initiated to address the root cause and prevent recurrence. If the complaint is isolated, the resolution is documented and the complaint is closed.
Complaint closure and trend monitoring
The complaint is formally closed with the complete investigation record, risk assessment, regulatory determination, and any CAPA references. Complaint data is aggregated for trend analysis, feeding into post-market surveillance and management review activities.
This process commonly relies on inputs such as the complaint report, device identification and lot data, manufacturing records, returned product examination results, and prior complaint history. It may be triggered by a customer report, a field service event, a distributor notification, or an internal quality observation. Connected systems often include quality management systems (QMS) like MasterControl, Veeva Vault, or ETQ, product lifecycle management systems, and regulatory submission platforms for MDR filing.
Key decision points include the initial risk classification that determines investigation priority and timeline, whether the investigation identifies a root cause tied to design, manufacturing, or labeling, whether the complaint meets MDR reporting criteria requiring regulatory submission, and whether the findings warrant a CAPA to address a systemic quality issue.
Complaints not logged promptly, delaying the start of investigation and risking regulatory reporting timeline compliance. Initial risk assessment underestimating severity, causing a reportable event to miss expedited investigation and filing deadlines. Incomplete investigation when returned product is not available or manufacturing records are difficult to retrieve, weakening root cause analysis. Regulatory reporting deadlines missed because the determination was delayed or the investigation was not prioritized. Complaint trends not identified because data is siloed in individual complaint records without systematic aggregation and analysis.
Orchestrates the full complaint lifecycle from intake through investigation, regulatory determination, and CAPA across customer-facing, quality, engineering, and regulatory teams in a single flow.
Routes complaints based on risk classification so potential reportable events are expedited while lower-risk issues follow standard timelines.
AI Agents parse complaint narratives at intake to flag potential adverse events, extract device identification data, and surface related prior complaints.
Tracks regulatory reporting timelines within the workflow to ensure MDR submissions are prepared and filed before deadlines.
Connects to QMS and product lifecycle platforms like MasterControl, Veeva Vault, and ETQ so complaint data, investigation records, and CAPA references are synchronized.
Preserves the complete complaint record including intake, risk assessment, investigation findings, regulatory determinations, and corrective actions for inspection readiness and post-market surveillance.