Quality assurance manager
Production manager
Regulatory affairs lead
Operations director
Supply chain manager
Compliance officer

This process is used when a deviation from defined quality specifications is identified during production, inspection, receiving, or any stage of the product lifecycle. It is triggered when materials fail incoming inspection, when in-process checks reveal out-of-specification conditions, when finished goods do not meet release criteria, or when customer complaints indicate a potential quality issue. Quality deviation approval is critical in manufacturing, pharmaceuticals, medical devices, food production, and any regulated industry where deviations must be formally assessed and dispositioned before work can continue.
The quality inspector or production operator identifies and documents the deviation. The quality assurance manager or quality engineer evaluates the deviation against acceptance criteria and risk impact. Production or operations management assesses the impact on schedules, inventory, and downstream processes. Regulatory affairs or compliance officers determine whether the deviation triggers reporting obligations or requires regulatory notification. For high-risk deviations, a material review board or executive quality authority may make the final disposition decision.
Faster deviation resolution by routing assessments to the right reviewers immediately with full context, reducing the time materials or production lines are on hold. Consistent disposition decisions because every deviation follows the same structured evaluation against defined acceptance criteria and risk thresholds. Reduced compliance risk through documented assessment, decision rationale, and corrective action tracking that satisfies regulatory and audit requirements. Lower scrap and rework costs because deviations are evaluated for use-as-is acceptability where appropriate, rather than defaulting to rejection without assessment. Stronger corrective action follow-through because CAPA requirements identified during deviation review are tracked to completion within the same process.
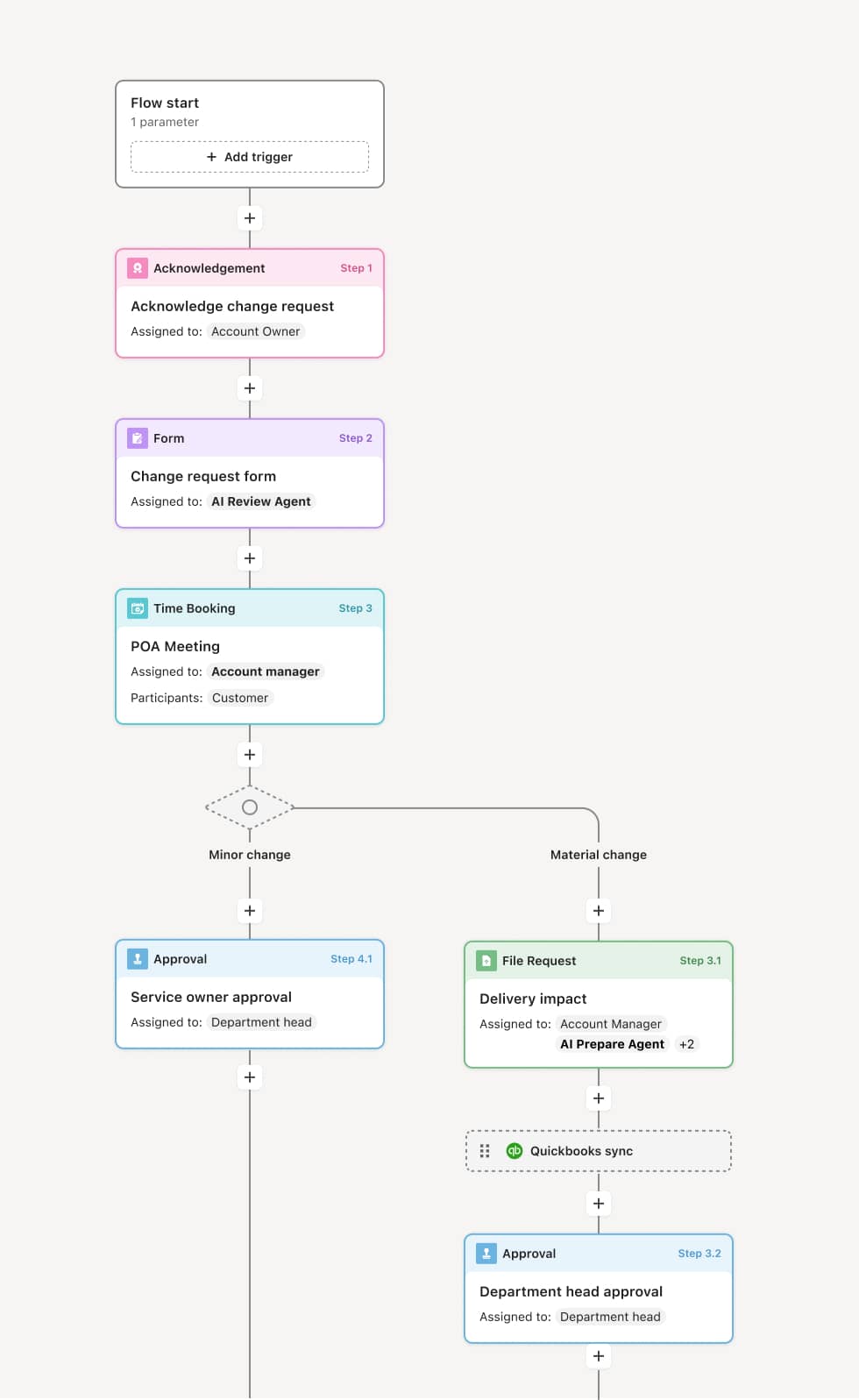
Your version of this process may vary based on roles, systems, data, and approval paths. Moxo’s flow builder can be configured with AI agents, conditional branching, dynamic data references, and sophisticated logic to match how your organization runs this workflow. The steps below illustrate one example.
Deviation identification and documentation
The process begins when a quality deviation is identified and formally documented. The deviation report includes a description of the nonconformance, the affected material or product, lot or batch numbers, the specification that was not met, and any immediate containment actions taken. An AI agent can assist by pulling relevant specification data, prior deviation history for the same material or process, and supplier records from connected quality systems to provide context for the assessment.
Risk assessment and impact evaluation
The quality team evaluates the deviation against risk criteria, including the severity of the nonconformance, its potential impact on product safety or efficacy, whether the deviation is isolated or systemic, and whether it affects downstream processes or customers. If the deviation is low-risk and falls within predefined acceptance criteria, it may be dispositioned quickly. If the risk is elevated—for example, affecting a regulated product or a customer-critical specification—the process branches to require a more comprehensive review, potentially involving regulatory affairs and a material review board.
Disposition decision
Based on the risk assessment, the authorized decision-maker determines the disposition of the affected material or product. Common dispositions include use-as-is with documented justification, rework to bring the material into specification, return to supplier, or scrap and disposal. The decision-maker must have the appropriate authority level for the risk category of the deviation. If corrective and preventive actions (CAPA) are required, they are initiated as part of this step and assigned to responsible owners with defined timelines. AI agents can surface relevant precedent decisions and regulatory guidance to support the disposition review.
Regulatory and compliance review
For deviations that may trigger regulatory reporting obligations—such as those affecting product safety, labeling, or batch release in regulated industries—the regulatory affairs or compliance team reviews the deviation and disposition to determine whether external notification or additional documentation is required. If regulatory action is needed, it is coordinated within the workflow to ensure timely and complete reporting.
Closure and corrective action tracking
Once the disposition is executed and any immediate actions are complete, the deviation is formally closed. If CAPA items were initiated, they remain tracked within the workflow until effectiveness verification confirms the root cause has been addressed. The complete deviation record—including the initial report, risk assessment, disposition decision, regulatory review, and CAPA outcomes—is retained as part of the quality management system.
This process commonly relies on inputs such as inspection reports, specification documents, batch or lot records, supplier quality data, and prior deviation history. It may be triggered by a failed inspection result, a production hold notification, a customer complaint, or a system alert from a quality management platform. Connected systems such as SAP QM, MasterControl, Veeva, or TrackWise provide quality and compliance data, while ERP systems like SAP or Oracle supply production and inventory context.
Key decision points include whether the deviation falls within predefined acceptance criteria for low-risk dispositions, whether the risk assessment indicates elevated impact requiring material review board involvement, what disposition is appropriate for the affected material or product, whether corrective and preventive actions are required, and whether the deviation triggers regulatory reporting obligations. If the deviation cannot be dispositioned at the initial review level, it is escalated to higher quality authority.
Deviations documented without sufficient detail, forcing quality reviewers to investigate before they can assess risk. Risk assessment criteria not consistently applied, leading to inconsistent disposition decisions across shifts or sites. Containment actions not taken promptly, allowing affected material to move downstream before the deviation is assessed. CAPA items initiated but not tracked to closure, leaving root causes unaddressed and increasing the likelihood of recurrence. Regulatory reporting timelines missed because the compliance review was not integrated into the deviation workflow.
Orchestrates deviation assessment across quality, operations, regulatory, and supply chain teams so that every deviation is evaluated by the right stakeholders with full context, without manual routing or email coordination.
AI agents assist with deviation context assembly by pulling specification data, prior deviation history, and supplier records from connected quality systems, reducing investigation time and ensuring completeness.
Routes deviations to the appropriate disposition authority based on risk category and product type, ensuring low-risk deviations are resolved quickly while high-risk cases receive material review board attention.
Connects to quality management and ERP systems such as SAP QM, MasterControl, or TrackWise to pull inspection data and push disposition outcomes back into the quality system of record.
Tracks corrective and preventive actions to closure within the same workflow, ensuring that CAPA commitments made during deviation review are followed through to effectiveness verification.