Drug safety officer
Principal investigator
Clinical research coordinator
Regulatory affairs director
Medical monitor
Pharmacovigilance manager

This process is used when a serious adverse event occurs in a clinical trial subject or is reported in association with a marketed product and meets the regulatory criteria for expedited reporting. It applies when the event must be assessed for seriousness, causality, and expectedness, and when reports must be submitted to the sponsor, IRB, and regulatory authorities within 24 hours for fatal or life-threatening events and 15 calendar days for other serious and unexpected events. Ideal for pharmaceutical sponsors, CROs, clinical trial sites, and marketing authorization holders.
The SAE reporting process typically involves the clinical investigator who identifies and reports the event, clinical research coordinators who document and submit the initial report, the sponsor’s safety team who assesses the event and prepares regulatory submissions, medical monitors who evaluate clinical significance and causality, and regulatory affairs who submit reports to health authorities and IRBs.
On-time regulatory submissions by managing the SAE from identification through submission within the mandated expedited timeline. Accurate seriousness and causality assessment because medical review is integrated into the reporting flow. Complete initial reports that include all available clinical details, reducing follow-up queries from sponsors and regulators. Protected subject safety through prompt assessment and communication of serious events to all responsible parties. Audit-ready SAE documentation that demonstrates regulatory compliance from event identification through submission and follow-up.
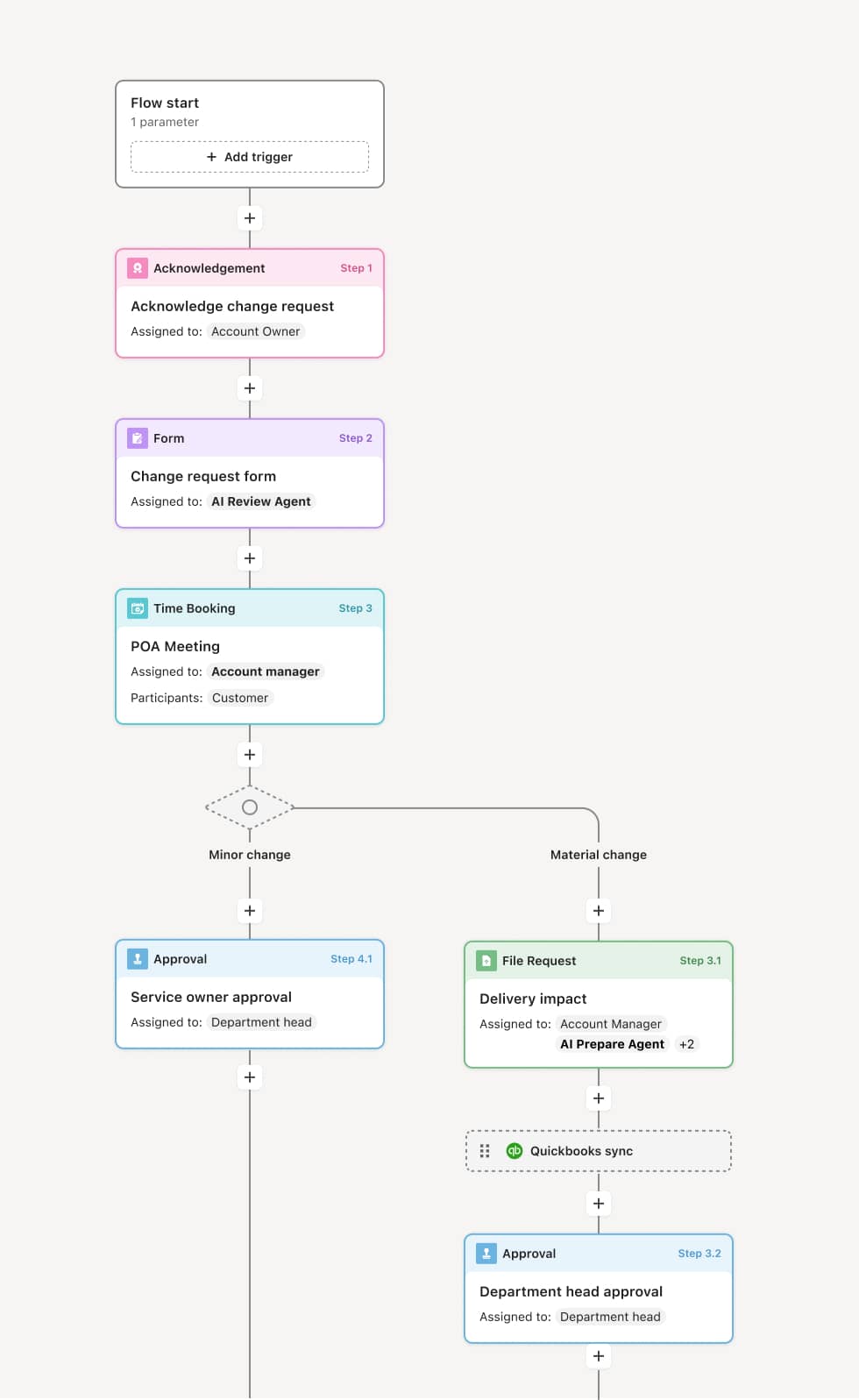
Your version of this process may vary based on roles, systems, data, and approval paths. Moxo’s flow builder can be configured with AI agents, conditional branching, dynamic data references, and sophisticated logic to match how your organization runs this workflow. The steps below illustrate one example.
SAE identification and initial notification
The process begins when the investigator identifies a serious adverse event in a clinical trial subject. The investigator or coordinator notifies the sponsor within 24 hours of becoming aware. For fatal or life-threatening events, immediate notification is required. An AI Agent can assist by pre-populating the SAE report form with the subject’s study data and verifying that all required fields are completed.
Detailed SAE documentation
The coordinator completes the detailed SAE report form including the event narrative, concomitant medications, relevant medical history, laboratory data, treatment provided, and event outcome. The investigator reviews and signs the report.
Sponsor safety assessment
The sponsor’s safety team receives the report and performs a medical assessment including seriousness confirmation, causality evaluation, and expectedness determination. If the event is both serious and unexpected (a SUSAR), expedited regulatory reporting is triggered. An AI Agent may compare the event against the investigator’s brochure to flag unexpected events.
Regulatory and IRB reporting
For SUSARs, the regulatory affairs team prepares and submits expedited reports to applicable health authorities within 7 calendar days for fatal or life-threatening events and 15 calendar days for other serious and unexpected events. The IRB is notified within the institution’s required timeframe.
Follow-up and case lifecycle
As additional information becomes available, follow-up reports are prepared and submitted. The case remains open until the event is resolved or all relevant information has been reported. The complete SAE record is preserved.
This process commonly relies on inputs such as the SAE report form, subject study data, concomitant medications, medical history, laboratory results, investigator’s brochure, and the study protocol. It may be triggered by an investigator’s identification of a serious event. Connected systems often include CTMS, EDC platforms like Medidata Rave, safety databases like Argus or ArisGlobal, and regulatory submission gateways.
Key decision points include confirmation that the event meets the regulatory definition of serious, the investigator’s and sponsor’s assessment of causality, whether the event is expected or unexpected based on the investigator’s brochure, and which regulatory authorities and IRBs require notification within what timeframes.
Initial investigator notification delayed beyond 24 hours, compressing the timeline for sponsor assessment and regulatory submission. Incomplete initial reports that require follow-up to obtain essential clinical details. Expectedness determination errors when the event is compared against an outdated version of the investigator’s brochure. Regulatory submission deadlines missed because assessment, preparation, and submission steps are not coordinated within the expedited timeline. Follow-up information not integrated into the case in a timely manner.
Orchestrates SAE reporting from identification through regulatory submission across investigators, coordinators, safety teams, and regulatory affairs within mandated expedited timelines.
Triggers immediate notification workflows when an SAE is identified, ensuring the sponsor is notified within 24 hours.
AI Agents pre-populate SAE report forms with subject study data and flag incomplete fields before submission.
Tracks regulatory submission deadlines — 7-day and 15-day timelines — within the workflow to ensure every required report is filed on time.
Connects to CTMS, EDC, and safety databases like Medidata, Argus, and ArisGlobal so subject data, safety assessments, and regulatory submissions are synchronized.
Preserves the complete SAE record including initial and follow-up reports, safety assessments, regulatory submissions, and IRB notifications for inspection and audit readiness.