Clinical operations manager
Principal investigator
Clinical research coordinator
Quality assurance auditor
Regulatory affairs specialist
Sponsor study director

This process is used when a deviation from the approved protocol, ICH-GCP guidelines, or institutional requirements is identified during the conduct of a clinical trial. It applies when the deviation involves missed visits, dosing errors, eligibility violations, consent process failures, specimen handling errors, or any other departure from the planned study conduct. It is common when site staff, monitors, principal investigators, quality teams, and sponsors must coordinate to document, assess, and resolve the deviation within regulatory expectations. Ideal for pharmaceutical sponsors, contract research organizations (CROs), academic medical centers, and clinical trial sites conducting trials under FDA, EMA, or other regulatory oversight.
The deviation management process typically involves clinical research coordinators who identify and document the deviation, principal investigators who assess the impact on subject safety and data integrity, clinical operations managers who oversee resolution and CAPA implementation, quality assurance auditors who review deviation trends and compliance, and sponsor study teams who are notified and assess impact on the overall trial.
Preserved trial data integrity because deviations are documented with their impact on data quality assessed and addressed. Protected subject safety through prompt assessment of whether the deviation affected or could affect participant well-being. Regulatory compliance with ICH-GCP, FDA, and IRB requirements for deviation documentation and reporting. Reduced recurrence through corrective and preventive actions tied to root cause analysis of significant deviations. Complete deviation records that satisfy sponsor audit, regulatory inspection, and IRB continuing review requirements.
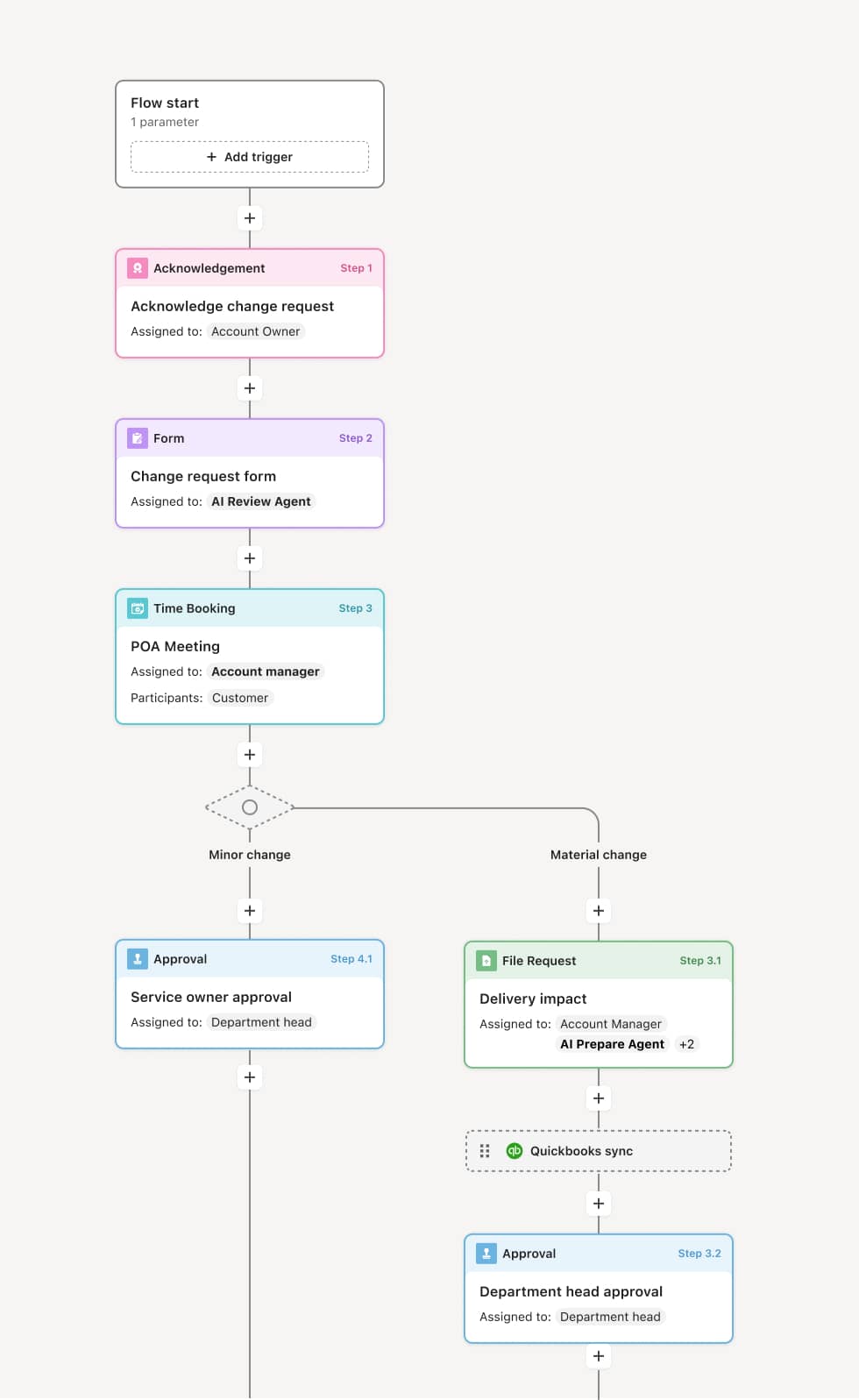
Your version of this process may vary based on roles, systems, data, and approval paths. Moxo’s flow builder can be configured with AI agents, conditional branching, dynamic data references, and sophisticated logic to match how your organization runs this workflow. The steps below illustrate one example.
Deviation identification and documentation
The process begins when a deviation is identified — by site staff during trial conduct, by a monitor during a site visit, or through data review. The deviation is documented with the description of what occurred, the protocol requirement that was not met, the date and circumstances, and any immediate corrective actions taken. An AI Agent can assist by classifying the deviation type and linking it to the specific protocol section that was violated.
Impact assessment
The principal investigator assesses the deviation’s impact on subject safety and data integrity. Deviations that affect or could affect participant safety, eligibility, or primary endpoint data are classified as major or critical. Minor deviations with no impact on safety or data integrity are classified accordingly. This classification determines the reporting and resolution requirements.
Notification and reporting
Major deviations are reported to the sponsor and, when required, to the IRB and regulatory authorities within specified timeframes. The sponsor’s study team is notified to assess impact on the overall trial. An AI Agent may prepare a deviation summary for sponsor and IRB reporting based on the documented details and impact assessment.
Root cause analysis and corrective action
For significant deviations, a root cause analysis is conducted to identify why the deviation occurred and what systemic factors contributed. Corrective and preventive actions (CAPAs) are developed, assigned, and tracked to implementation. CAPAs may include additional staff training, protocol amendments, process changes, or system modifications.
Resolution and verification
The CAPA implementation is verified for effectiveness. The deviation is formally resolved and closed with the complete record of the event, assessment, corrective actions, and verification. If the CAPA addresses a systemic issue, its effectiveness is monitored over subsequent trial activities.
Trend analysis and quality oversight
Deviation data is aggregated for trend analysis — by deviation type, site, protocol, and root cause. Trends inform quality oversight activities, site management decisions, and protocol amendment considerations.
This process commonly relies on inputs such as the deviation report, protocol and amendment documents, subject records, monitoring reports, and training documentation. It may be triggered by site staff observation, monitoring visit findings, data management queries, or audit observations. Connected systems often include clinical trial management systems (CTMS) like Medidata or Veeva Vault, electronic data capture (EDC) systems, and quality management systems for CAPA tracking.
Key decision points include the classification of the deviation as minor, major, or critical based on its impact on subject safety and data integrity, whether the deviation requires reporting to the sponsor, IRB, or regulatory authorities, what corrective and preventive actions are needed to address the root cause, and whether the deviation affects the subject’s continued participation or the validity of collected data.
Deviations not identified promptly, delaying documentation and potentially compounding the impact on data integrity. Impact assessment underestimating significance, causing major deviations to be classified as minor and not reported appropriately. CAPA not addressing root cause, resulting in recurrence of the same or similar deviations. Sponsor and IRB notifications delayed beyond required reporting timelines, creating compliance findings. Deviation data not aggregated for trend analysis, missing patterns that indicate systemic site or protocol issues.
Orchestrates deviation management from identification through resolution across site staff, investigators, sponsors, and quality teams in a single coordinated flow.
Routes deviations based on severity classification so major deviations receive immediate attention and reporting while minor deviations follow standard timelines.
AI Agents classify deviation types and link them to protocol requirements at documentation, accelerating the initial assessment and ensuring completeness.
Tracks CAPA implementation and verification within the workflow so corrective actions are assigned, completed, and confirmed as effective.
Connects to CTMS and EDC platforms like Medidata and Veeva Vault so deviation records are synchronized with trial conduct and subject data.
Captures deviation trends across sites, protocols, and deviation types, enabling quality oversight and proactive risk management for the trial program.