Clinical operations director
Research ethics coordinator
Surgical services manager
Principal investigator
Patient experience officer
Compliance officer

This process is used before any medical procedure, treatment, or research participation that requires the patient’s or participant’s informed authorization. It applies when disclosure of risks, benefits, alternatives, and the right to refuse must be communicated and documented, and when regulatory or institutional requirements dictate specific consent procedures. It is common in surgical pre-operative workflows, clinical trial enrollment, diagnostic procedures with material risk, and any clinical scenario where the patient’s or participant’s autonomous decision-making must be supported and recorded. Ideal for hospitals, surgical centers, clinical research sites, behavioral health facilities, and any healthcare organization managing consent processes.
The informed consent process typically involves the treating physician or principal investigator who provides disclosure and answers questions, clinical coordinators or research coordinators who facilitate the consent workflow and documentation, the patient or participant who receives information and provides authorization, witnesses or interpreters who participate when required, and compliance or quality teams who oversee consent process standards.
Legally and ethically valid consent because the process ensures adequate disclosure, comprehension verification, and voluntary authorization are documented. Reduced consent-related compliance findings through structured workflows that ensure the correct document version, required signatories, and proper timing. Better patient or participant understanding by providing structured time for questions and comprehension verification within the workflow. Complete consent documentation immediately available in the clinical or research record, preventing downstream findings during audits, monitoring, or litigation. Consistent consent practices across providers, departments, and study sites, regardless of the clinical or research context.
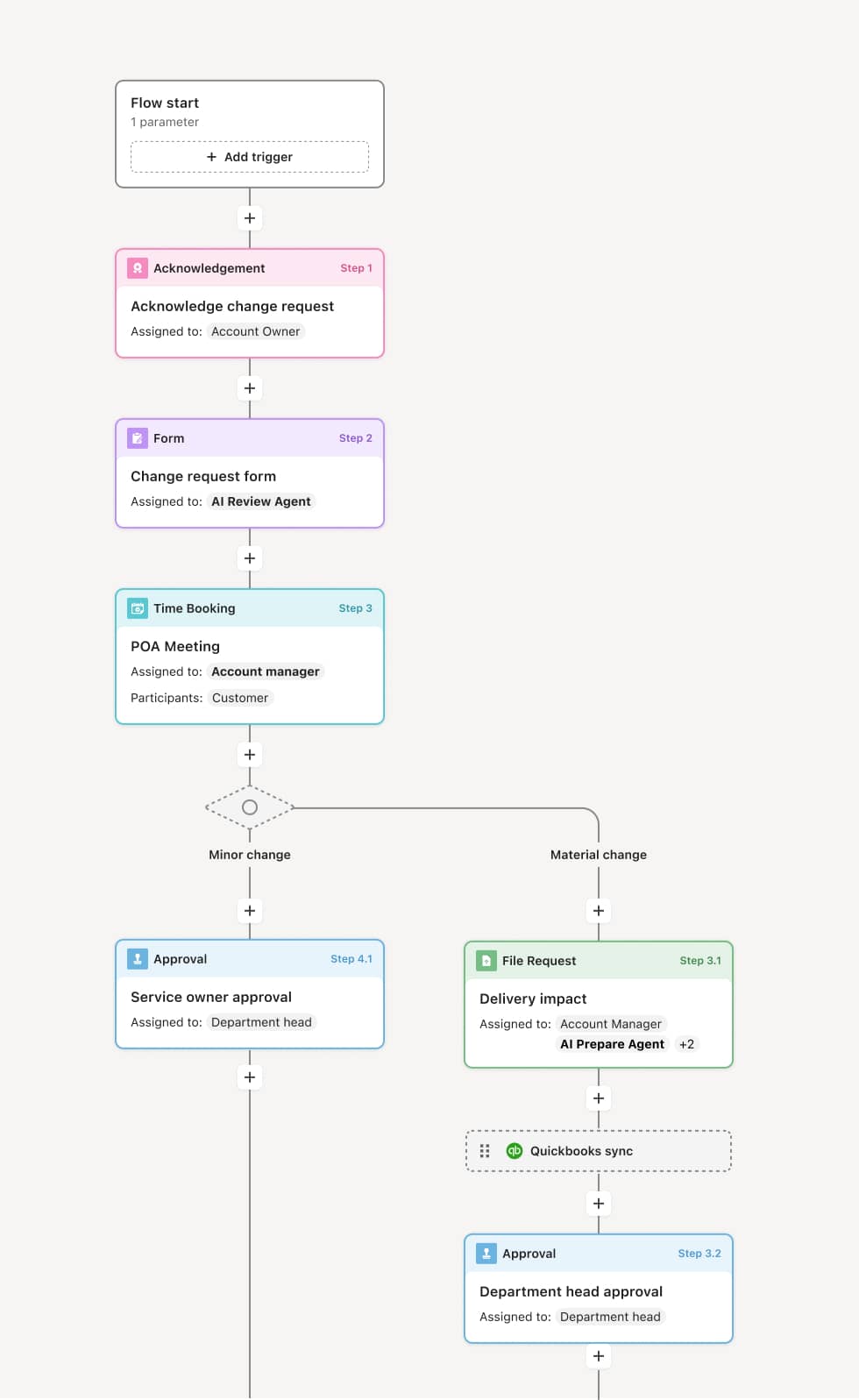
Your version of this process may vary based on roles, systems, data, and approval paths. Moxo’s flow builder can be configured with AI agents, conditional branching, dynamic data references, and sophisticated logic to match how your organization runs this workflow. The steps below illustrate one example.
Consent preparation
The process begins when a provider or coordinator identifies that informed consent is required for a scheduled procedure, treatment, or research activity. The correct consent document is retrieved — matching the procedure, the current approved version, and the patient’s language preference. An AI Agent can assist by verifying that the document version is current, that it matches the planned procedure or study, and that any required translations or supplementary materials are included.
Disclosure and discussion
The treating physician or principal investigator provides the patient or participant with a clear explanation of the proposed procedure or study, including the nature of the intervention, expected benefits, material risks, alternatives, and the right to refuse or withdraw. The discussion is conducted in a setting that supports understanding, with adequate time for questions. If an interpreter is needed, their participation is arranged and documented.
Comprehension verification
Before signing, the coordinator or provider confirms that the patient or participant understands the disclosed information. This may involve asking the patient to summarize key points, answering follow-up questions, or using comprehension assessment tools — particularly for research studies or complex procedures. If comprehension is not adequate, additional explanation is provided.
Signature and documentation
The patient or participant signs the consent document. The provider and any required witnesses also sign. The consent document is date- and time-stamped. For research studies, the signed consent is recorded in both the participant’s study file and the regulatory binder. For clinical care, the document is entered into the medical record. An AI Agent may verify that all required signatures, dates, and witness fields are completed before the document is finalized.
Record preservation and accessibility
The signed consent document is stored in the appropriate system — the EHR for clinical consent, the regulatory file for research consent. A copy is provided to the patient or participant. The consent record is linked to the procedure, treatment, or study enrollment so it is immediately accessible during care delivery, monitoring, or audit.
Re-consent triggers
If the procedure or study changes materially — such as a protocol amendment, new risk information, or a change in the planned intervention — the consent process is reinitiated with updated disclosure and a new consent document.
This process commonly relies on inputs such as the consent document (procedure-specific or study-specific), patient or participant identification, procedure or study details, interpreter availability, and any supplementary educational materials. It may be triggered by a procedure scheduling event, a research enrollment workflow, or a treatment plan change. Connected systems often include EHR platforms like Epic or Cerner for clinical consent documentation, electronic consent (eConsent) platforms for research, and document management systems for version control.
Key decision points include whether the correct and current version of the consent document is being used, whether the patient or participant demonstrates adequate comprehension of the disclosed information, whether all required signatories have completed the consent documentation, and whether any changes to the procedure or study require re-consent with updated disclosure.
Outdated consent document versions used, creating regulatory and legal findings. Consent obtained without adequate time for patient questions, undermining the voluntary and informed nature of the authorization. Missing signatures, dates, or witness requirements that invalidate the consent documentation. Consent not documented in the medical or research record before the procedure or study activity begins. Re-consent not triggered when material changes occur to the planned intervention or study protocol.
Orchestrates the informed consent workflow from document preparation through signature and record preservation across providers, coordinators, and patients or participants.
Ensures the correct consent document version is used by managing version control and linking documents to the specific procedure or study.
AI Agents verify document completeness at the point of signing, flagging missing signatures, dates, or required fields before the document is finalized.
Supports comprehension verification by structuring the consent workflow to include time for discussion, questions, and confirmation before signature.
Connects to EHR and eConsent platforms like Epic, Cerner, and research eConsent tools so signed documents are immediately stored in the appropriate record system.
Preserves the complete consent record and triggers re-consent when material changes occur, ensuring ongoing compliance with regulatory, institutional, and ethical requirements.